癌の「遺伝子調節化学予防」と「遺伝子調節化学療法」
がんの「遺伝子調節化学療法」と「遺伝子調節化学予防」から「RB再活性化スクリーニング」の発想に至った経緯
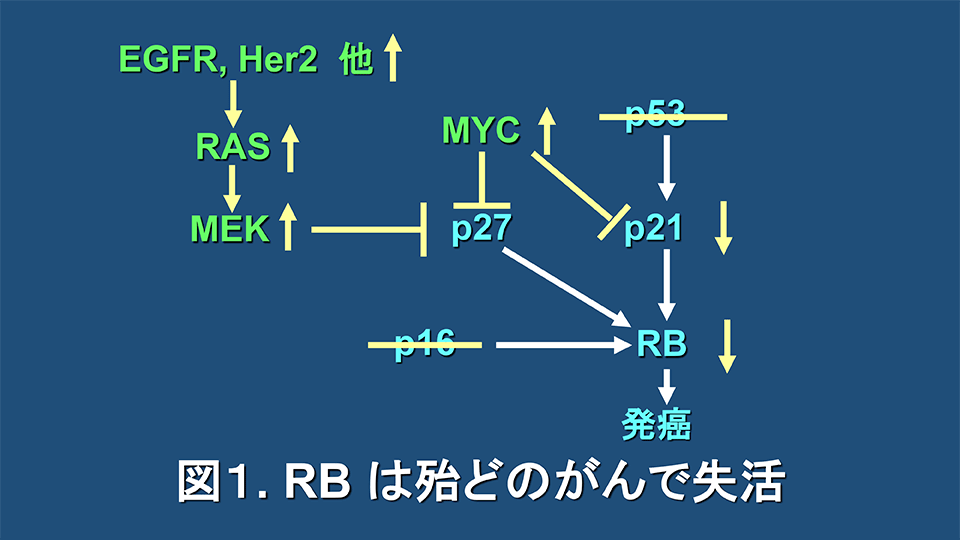
酒井は大学院の時代から京都府庁衛生部に勤めていた頃までは、本学生化学教室におられた西野輔翼先生や、京都大学の成宮周先生や、当時愛知県がんセンターにおられた福島雅典先生のご指導の下に、抗腫瘍性プロスタグランディンの研究に従事していた(Cancer Lett. 1983; 17: 289-94, Prostaglandins. 1984; 27: 17-26, Cancer Res. 1989; 49: 1193-6, FEBS Lett. 1990; 270: 15-8, Biochem Biophys Res Commun. 1991; 179: 1662-9)。これらの仕事の中で、Δ12-prostaglandin J2が、処理後数時間でN-mycの発現を強力に抑制し、その後G1期停止作用を示した結果は(FEBS Lett. 1990; 270: 15-8)、その後の私達の研究に大きな影響を与えることとなった。この実験を計画したのは、当時からMYCはG1期を促進させることが知られていたので、Δ12-prostaglandin J2がN-mycの発現を抑制することによりG1期停止を起こすことが可能ではないかと想定したからであった。今では、図1に示すように、MYCはサイクリン依存性キナーゼ(CDK)阻害因子のp27やp21を抑制することによりRBをリン酸化型(不活性型)にしてG1期を促進させることが明らかにされているので、Δ12-prostaglandin J2がN-mycの発現を抑制することによりRBを活性化型にしてG1期停止を引き起こしていたことが強く推定される。
しかしながら、当時はRBの機能も明らかでなく、そこまでは予測できていなかった。ただし、Δ12-prostaglandin J2という低分子化合物がN-mycの発現を抑制することにより、言い換えればN-mycの遺伝子発現の「量」を調節することにより、G1期停止を起こすことを示唆できたことから、その後、低分子化合物により、遺伝子発現を調節することでがんの治療や予防を目指す、「遺伝子調節化学療法」や「遺伝子調節化学予防」の研究、さらには、それより創案された「RB再活性化スクリーニング」の発想の端緒となった。
その頃、酒井は、福島雅典先生のご助言で、10年間のNature、Scienceを読了して、今後進むべき方向をがん抑制遺伝子の研究に決めた。そこで、最初のがん抑制遺伝子をクローニングした、ハーバード医科大学眼科のDr. Thaddeus P. Dryjaのラボに留学した。いくつかの研究室の中からDryjaのラボを選択した大きな理由は、Dryjaのラボでは、RB遺伝子プロモーターの研究もさせていただけるということだった。すなわち、がん抑制遺伝子のプロモーターの研究の延長には、Δ12-prostaglandin J2がN-mycの発現を抑制することによる治療法のように、低分子化合物によりプロモーターレベルでがん抑制遺伝子の発現を促進させることによる新しいがん治療法やがん予防法の可能性が開けることを期待したからである。
留学時の成果とそれに引き続く創薬への経緯に関しては、「trametinibの発見の歴史と近況」の項に述べたので、ここでは、その要約と追加情報を述べることとする。留学時には、RB遺伝子プロモーターの点突然変異からの失活による網膜芽細胞腫家系の発見と(Nature. 1991; 353: 83-6, 図2)、RBプロモーターに過剰メチル化が存在する網膜芽細胞腫を見いだした(Am J Hum Genet. 1991; 48: 880-8, 図2)。
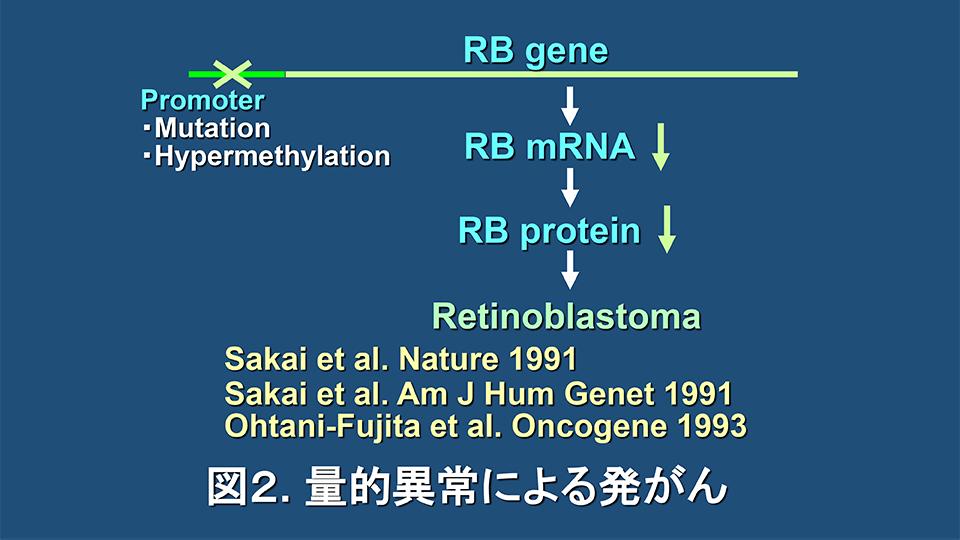
ただし、当時の分子生物学の教科書では、遺伝子プロモーターの過剰メチル化は、遺伝子発現の失活の原因というよりは、遺伝子発現の失活の結果ではないかという考え方の方が支配的であった。そこで、帰国後、Dryjaとは独立に研究を行い、RB遺伝子プロモーターの過剰メチル化により、プロモーター活性が激減することを証明し、報告した(Oncogene. 1993; 8: 1063-7, 図2)。これらの研究の意義として、発がんは遺伝子発現の量的異常でおきることを明らかにした点と、がん抑制遺伝子が過剰メチル化により失活することを初めて証明したことから、その後の「がんとエピジェネティクス」研究の発展につながったことなどがあげられる。重要なのは、このことにより、発がんの本質が遺伝子発現の異常であれば、その遺伝子発現を正常化することにより、がんを治療したり診断したりすることが可能ではないかという思いに至ったことである。
帰国後の1993年に、大谷直子(現東京理科大教授)、曽和義広(現准教授)らが大学院生として在籍していた。その時の抄読会で、がん抑制遺伝子p53が転写因子として、CDK阻害因子のp21/WAF1/Cip1(以下p21)の発現を増強することにより、RBを活性化型(脱リン酸化型)にして、がん細胞の増殖を抑制する経路が紹介された(Cell. 1993; 75: 817-25, Cell. 1993; 75: 805-16)。その頃、p53遺伝子が突然変異などで失活していて発がんに至る場合に、p53遺伝子を遺伝子療法でがん細胞に導入する治療法が注目されていたが、酒井は遺伝子導入の効率など考えると、実現性は低いと考え、他の方法を模索していた。そこで、図3に示すように、p53遺伝子が突然変異で失活しているがん細胞において、p21の発現が低下していても、薬剤でp21の発現を上昇させれば、RBが正常型に変換され、がん細胞の増殖を抑制できるのではないかと考えた。
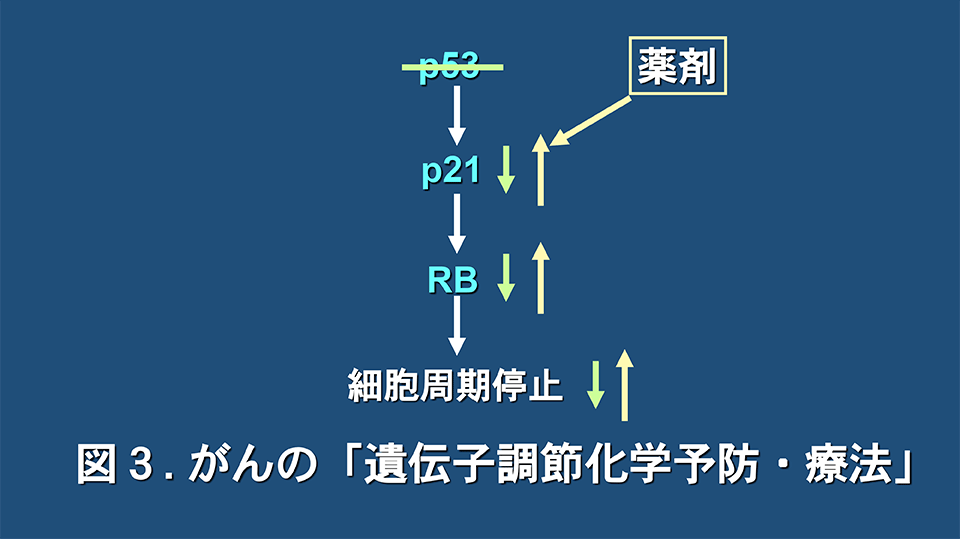
すなわち、p53遺伝子が突然変異で失活しても低分子化合物の投与でp53遺伝子の機能を代償できないかと考えた訳である。このような治療法や予防法を「遺伝子調節化学療法」、或いは「遺伝子調節化学予防」と名付けることとした(Jpn J Hyg. 1996; 50: 1036-46, Biofactors 2000; 12: 283-7)。
最初に見いだしたp21誘導物質は、食物繊維の代謝物質であり、がん予防成分と考えられていた酪酸であった。酪酸はp21をプロモーターのSp1部位を介して活性化し、RBを脱リン酸化型にすることにより、細胞周期をG1期(Rポイント)で停止させた(J Biol Chem. 1997; 272: 22199-206)。次に酪酸はHDAC阻害作用も有するので、HDAC阻害剤でも検討したところ、やはり同様に、同じSp1部位を介してp21をプロモーターレベルで活性化し、RBを脱リン酸化型にして細胞周期をG1期で停止させた(Biochem Biophys Res Commun. 1997; 241: 142-50)。
また、p21だけでなく、他のp53により誘導される遺伝子として、アポトーシスを誘導するDR5や遺伝子修復を行うgadd45などがあるが、これらの遺伝子発現を増強させることにより、失われたp53の機能を代償できる物質を検索した結果、HDAC阻害剤がこれらの遺伝子の発現を増強させることを見いだした(Oncogene. 2004; 23: 6261-71, Oncogene. 2003; 22: 7762-73)。特にDR5の発現増強物質はその後数多く見いだすことができたが、それに関しては、「癌の「分子標的併用療法」及び「分子標的併用予防」」の項目を参照されたい。
また一方では、ヒトがんの約半数で失活するとされる代表的がん抑制遺伝子のp53やp16が失活する際に、RBはタンパク質レベルで失活する(図1)。加えて、代表的がん遺伝子であるMYCの活性化だけでなく、細胞膜に存在するEGFR他の多くのがん遺伝子の活性化や、その下流のRAS-RAF-MEK経路の活性化などによってもRBタンパク質は失活する(図1)。今では、これら以外にも種々の発がん経路の下流にRBタンパク質の失活が位置していることが知られている。
したがって、種々のがん分子標的薬は、最終的にRBをタンパク質レベルで再活性化することが想定される。そのため、RBを活性化型にするCDK阻害因子の発現を上昇させる物質のスクリーニングや、逆にRBを失活させるサイクリンの発現を抑制する物質をスクリーニングできれば、新規がん分子標的薬を発見することができる可能性が考えられた。そこで、このようなスクリーニング法を総称して、「RB再活性化スクリーニング」と名付けることとした。
そこで、いくつかの製薬会社とこれらのスクリーニングを行った結果、三種類の臨床試験に入った薬剤を見いだしたが、その成果に関しても「trametinibの発見の歴史と近況」の項に述べたので割愛する。ここでは、一点興味深い点のみを指摘することとする。代表的がん抑制遺伝子でCDK阻害因子であるp16のファミリー分子であるp15の発現を上昇させる物質のスクリーニングをJT医薬総合研究所と行った結果、進行性BRAF変異メラノーマ患者に対してfirst-in-classのMEK阻害剤として日米欧で承認されたtrametinib/トラメチニブ(商品名Mekinist/メキニスト)を共同で発見した訳であるが、元々は、ヒトがんの約半数で失活するとされるp16が失活している場合に、そのファミリーであるp15の発現を上昇させることにより、p16の機能を代償しうる薬剤を見いだすことを期待していた。その結果得られたtrametinibは進行性BRAF変異メラノーマ患者を対象に承認された訳だが、p16は遺伝性メラノーマ家系の原因遺伝子でもあり、かつメラノーマで高頻度に失活することから、当初の期待通りに、失活したp16の機能を代償している可能性も十分考えられる。
以上、抗腫瘍性プロスタグランディンの研究の経験から得られた、「低分子化合物で発がんに関与する遺伝子発現の調節が治療や予防に使えるかも知れない」という発想が、RB遺伝子プロモーターの研究からさらにその意を強くし、独立したラボを持ってから、遺伝子調節化学療法、或いは遺伝子調節化学予防法を創案し、そこから、RB再活性化スクリーニング法を確立し、創薬に至ることができた訳である。現在はこのノウハウをさらに発展させて、他の分子標的薬の開発を行っている。
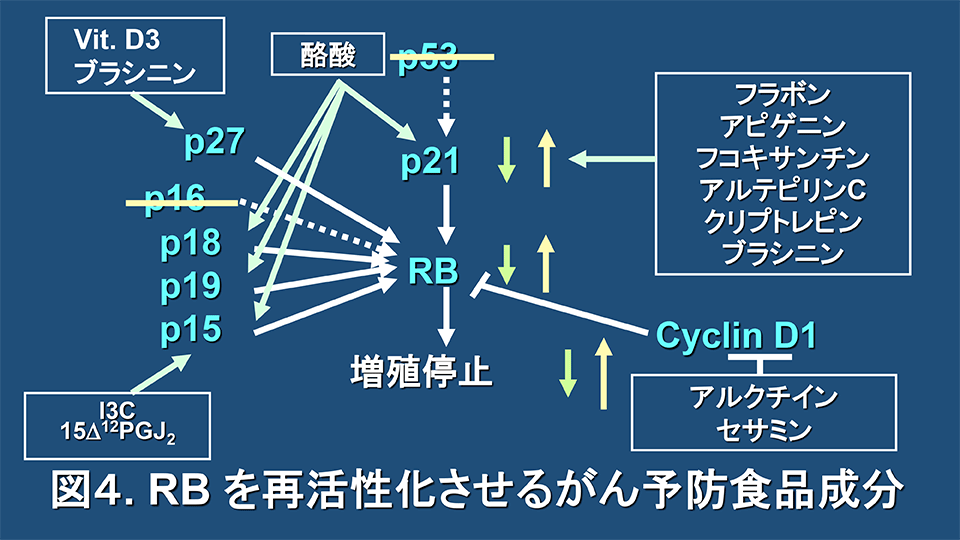
一方、がん予防もRB再活性化が関与している可能性を考え、多くのがん予防食品成分やがん予防物質を用いて検証したところ、数多くのがん予防食品成分やがん予防物質が、極めて興味深いことに最終的にRBをタンパク質レベルで再活性化していることを明らかにした(図4)。これらの論文は多数にわたるのでここでは省略するが、現在、某大手食品企業とRBを再活性化する「究極のがん予防ジュース」の開発研究も行っている。
初期の抗腫瘍性プロスタグランディンの研究
Sakai T, Yamaguchi N, Kawai K, Nishino H, Iwashima A.
Prostaglandin D2 inhibits the proliferation of human neuroblastoma cells.
Cancer Lett. 1983; 17: 289-94.
Sakai T, Yamaguchi N, Shiroko Y, Sekiguchi M, Fujii G, Nishino H.
Prostaglandin D2 inhibits the proliferation of human malignant tumor cells.
Prostaglandins. 1984; 27: 17-26.
Sakai T, Aoike A, Marui N, Kawai K, Nishino H, Fukushima M.
Protection by cycloheximide against cytotoxicity induced by vincristine, colchicine, or Δ12-prostaglandin J2 on human osteosarcoma cells.
Cancer Res. 1989; 49: 1193-6.
Marui N, Sakai T, Hosokawa N, Yoshida M, Aoike A, Kawai K, Nishino H, Fukushima M.
N-myc suppression and cell cycle arrest at G1 phase by prostaglandins.
FEBS Lett. 1990; 270: 15-8.
Marui N, Nishino H, Sakai T, Aoike A, Kawai K, Fukushima M.
Δ12-prostaglandin J2 mimics heat shock in inducing cell cycle arrest at G1 phase.
Biochem Biophys Res Commun. 1991; 179: 1662-9.
RB遺伝子プロモーターに点突然変異や過剰メチル化を見いだした論文
Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP.
Oncogenic germ-line mutations in Sp1 and ATF sites in the human retinoblastoma gene.
Nature. 1991; 353: 83-6.
Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP.
Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene.
Am J Hum Genet. 1991; 48: 880-8.
RB遺伝子プロモーターが過剰メチル化で失活することを明らかにした論文
Ohtani-Fujita N, Fujita T, Aoike A, Osifchin NE, Robbins PD, Sakai T.
CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene.
Oncogene. 1993; 8: 1063-7.
がんの遺伝子調節化学予防、あるいは遺伝子調節化学療法を思いつくきっかけになった論文
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B.
WAF1, a potential mediator of p53 tumor suppression.
Cell. 1993; 75: 817-25.
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ.
The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases.
Cell. 1993; 75: 805-16.
遺伝子調節化学療法、或いは遺伝子調節化学予防を提唱した論文
Sakai T
Molecular cancer epidemiology-The present status and future possibilities-
Jpn J Hyg. 1996; 50: 1036-46.
Sowa Y, Sakai T.
Butyrate as a model for “gene-regulating chemoprevention and chemotherapy”.
Biofactors 2000; 12: 283-7.
私達が酪酸やHDAC阻害剤が遺伝子調節化学予防や遺伝子調節化学療法の最初のモデルとして報告した論文
Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, Fujita T, Ohtani-Fujita N, Matsukawa Y, Tokino T, Yamagishi H, Oka T, Nomura H, Sakai T.
Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line.
J Biol Chem. 1997; 272: 22199-206.
Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites.
Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, Sakai T.
Biochem Biophys Res Commun. 1997; 241: 142-50.
p53遺伝子の下流遺伝子であるDR5やgadd45の発現をHDAC阻害剤が増強させることを見いだした論文
Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T.
Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells.
Oncogene. 2004; 23: 6261-71.
Hirose T, Sowa Y, Takahashi S, Saito S, Yasuda C, Shindo N, Furuichi K, Sakai T.
p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y.
Oncogene. 2003; 22: 7762-73.